...
Phenotypic Consequences of Karyotype Variation

Our lab focuses on understanding how large-scale chromosome changes, known as aneuploidy and polyploidy, drive cancer evolution and adaptation. While most normal human cells maintain a strict diploid state (two copies of each chromosome), cancer cells are often characterized by aneuploidy, which involves gains, losses, or rearrangements of entire chromosomes. This chromosomal instability (CIN) reshapes gene dosage and significantly alters cell behavior, contributing to tumor progression and therapy resistance. Approximately 88% of cancers exhibit aneuploidy, often from the earliest stages, and experimental correction of these karyotypes typically reduces cancer cell fitness, highlighting the tumor's dependence on this altered state.
The central mission of our research is to move beyond simply detecting CIN to predicting how karyotypes evolve and shape the cellular phenotype (observable characteristics like growth rate, death rate, metabolic state, etc.) over time and in different environments. We tackle this challenge by integrating mathematical modeling, single-cell sequencing, spatial transcriptomics, and metabolic profiling. A key advancement led by our team extended foundational mathematical models of chromosome mis-segregation (MS). While earlier models were limited to one chromosome at a time, our work enabled modeling MS across the entire set of chromosomes (Kimmel & Beck, 2023). This allowed us to derive karyotype transition probabilities for any given CIN rate. Because the full transition matrix is immense (effectively 70×10^18 by 70×10^18 states) and cannot be stored, our team derived the necessary equations to compute these probabilities empirically (Beck & Andor, 2024 preprint). This foundational modeling capability underpins our efforts to transform insights into karyotype-to-phenotype relationships into actionable strategies for controlling tumor progression.
We have developed several integrative tools to map karyotype changes to their functional consequences:
ALFA-K (Local Adaptive Mapping of Karyotype Fitness Landscapes): This tool predicts the fitness landscape of karyotypes using time-series single-cell data. By analyzing longitudinal data (tracking changes over time), ALFA-K can estimate the relative fitness of not only observed karyotypes but also thousands of related, unobserved karyotypes that are just one or two mis-segregation events away. This allows us to predict karyotype evolution. Key insights from ALFA-K include: • The fitness effect of gaining or losing a chromosome depends heavily on the existing karyotype context; divergent karyotypes respond differently to the same chromosomal change. • Environmental pressures (like cisplatin treatment) and genomic events (like Whole Genome Doubling - WGD) significantly reshape the karyotype fitness landscape, influencing which karyotypes are selected. WGD appears to increase access to advantageous karyotypes, enhancing adaptability. • The rate of chromosome mis-segregation itself can alter which karyotype becomes dominant, modulating the trade-off between a karyotype's intrinsic fitness and its robustness (e.g., lower-ploidy karyotypes might be favored at high mis-segregation rates despite lower fitness).

ALFA-K workflow & application. Longitudinal single-cell DNA-seq (A) informs a three-step pipeline for karyotype-fitness landscapes inference (B): 1.Observed karyotypes receive empirical fitness scores; 2. fitness for all von-Neumann neighbours (±1 chromosome) is estimated; 3. Gaussian-process interpolation completes the landscape. Validation withholding the final 25 days shows ALFA-K (purple) predicts forward dynamics better than a “no-evolution” baseline (grey), even when adaptation is nearly complete. (C) Application example. TNBC time-course with six sampling points (red) yields 50 input karyotypes. ALFA-K expands these to ~2,000 nearby states (<2 mis-segregations apart) and projects fitness onto UMAP. Input constitutes < 3 % of landscape karyotypes and ~10⁻¹⁴ % of total theoretical space.
S3MB (Stochastic State-Space Model of the Brain): This population-based model simulates glioblastoma (GBM) growth within the brain environment, linking cell-scale dynamics to observable tumor growth patterns at millimeter resolution. It incorporates factors like resource availability (oxygen, glucose), tissue stiffness, vasculature, and treatment effects. Using S3MB, we found that higher-ploidy GBM cells switch to glycolysis more readily (i.e., at higher oxygen thresholds) than lower-ploidy cells (Cancer Research 2025). Indeed we found a strong correlation between the oxygen levels in normal tissues and the frequency of WGD in tumors arising from those tissues, supporting the role of oxygen availability in shaping ploidy evolution
We are also developing more sophisticated models of the brain microenvironment, incorporating the crucial role of astrocytes in regulating energy flow, to understand how GBM cell karyotypes might influence infiltration patterns and reliance on normal brain circuits.
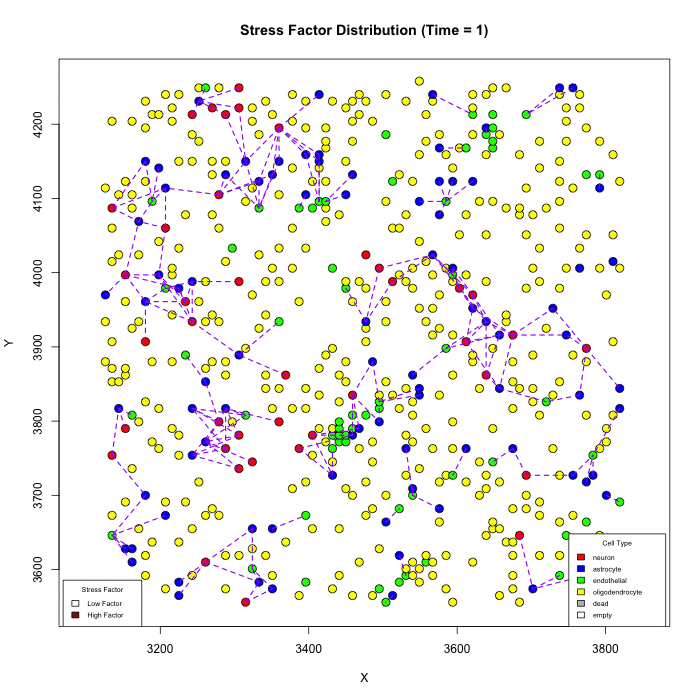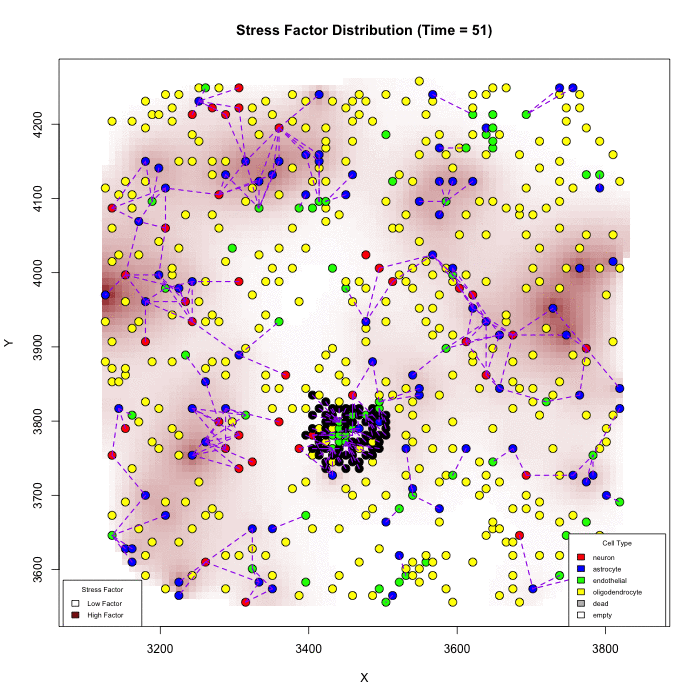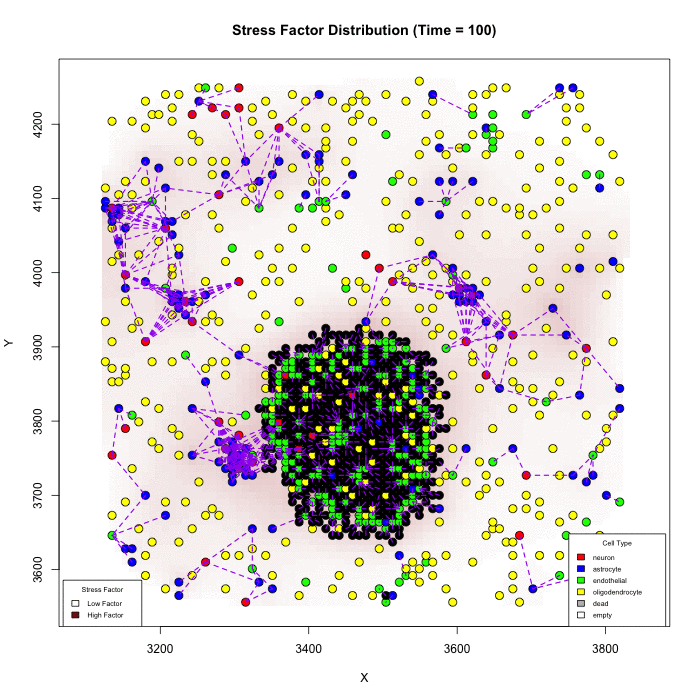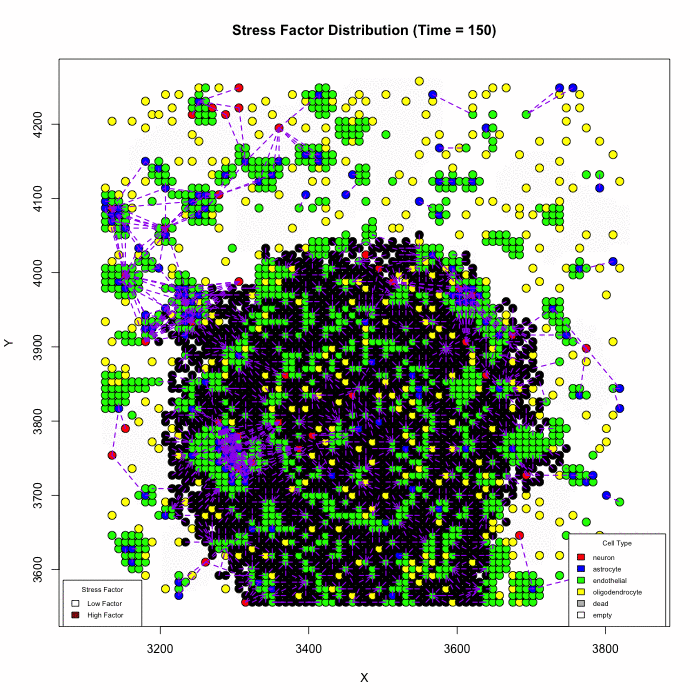

Does Karyotype Shape GBM Infiltration Patterns? Simulations begin with identical parameters except for angiogenic-stress signaling. Left: cancer astrocytes secrete their own stress factor, sustaining compact growth. Right: only normal astrocytes supply the factor, forcing malignant clones to infiltrate neural circuits, yielding diffuse spread. Panels illustrate spatial distributions after equivalent simulated time.
.CLONEID: Recognizing that time is crucial for understanding cancer adaptation, we developed CLONEID, a software framework and database to integrate and track genotypic (karyotype, mutations, transcriptional changes) and phenotypic (e.g., morphology, growth rate) data from cell lineages over time during experiments. It allows us to connect pedigree information, microscopy images, and sequencing data.

Phylogram of the gastric cancer cell line MKN-45, with 23 lineages. Phylograms in our CLONEID database each have between 2 - 2726 lineages, accompanied by high-quality images and segmentation masks.
This real-time monitoring enables us to:
• Observe adaptation directly, for instance, tracking how growth rates change under nutrient deprivation (like low glucose or phosphate) and correlating this with specific karyotype changes. We've observed parallel evolution, where the same advantageous karyotype emerges independently in replicate experiments under the same selective pressure.
• Generate and characterize isogenic cell lines (genetically identical except for ploidy) to study the specific effects of WGD on drug sensitivity. Preliminary findings suggest high-ploidy cells may be more sensitive to targeted therapies but resistant to cytotoxic drugs, while near-diploid cells show the opposite pattern.
• Build a rich, multi-modal dataset linking genotype, phenotype, and lineage history across thousands of lineages and tens of thousands of single cells, facilitating collaborations on topics like image segmentation of rare cell types.
CLONEID web portal for lineage-resolved multi-omics. Dashboard shows a time-stamped lineage tree for the SNU-668 gastric cancer cell lines. Blue squares mark nodes with single-cell karyotype heat-maps; red squares indicate linked morphology datasets. Users can zoom from root to tip to export paired genotype-phenotype snapshots—providing longitudinal inputs for models such as ALFA-K.
The long-term vision for CLONEID is to establish an infrastructure for collecting and connecting genotypic and phenotypic experimental data across labs worldwide, enhancing the value and interpretability of genomic data.
...
Characterizing cytotoxic therapy induced shifts in the cost-to-benefit ratio of high ploidy
. We previously coined the “tip-over hypothesis of DNA damage therapy sensitivity”, proposing that cytotoxic therapy is effective if it pushes a cell’s somatic copy number alteration (SCNA) load above a tipping point. Variable proximity of co-existing tumor cells to this tipping point imply that dose-response relations need not be monotonic. We test the potential of tumor cell DNA content and dNTP substrate availability to predict a tumor’s vulnerability to increased SCNA rates (e.g. due to cytotoxic therapy).

Opposing selective forces explain why cancer therapies shift the Goldilocks zone of tumor aneuploidy. We distinguish cells along two dimensions: their SCNA load (grayscale, as in A) and their ploidy (drawn as cell size). (A) The Goldilocks zone of tumor aneuploidy lies at intermediate SCNA loads among therapy-naïve patients (-; blue arrow). For patients exposed to chemo/radiation therapy (+; red arrow), intermediate and high SCNA loads have a similar risk of disease progression. (B) Interaction between two selective pressures – energy scarcity and chromosomal instability (CIN) – can explain differences between therapy-naïve and therapy-exposed patients. Compared to low ploidy cells, high ploidy cells are more likely to survive mutations and accumulate high SCNA loads (y-axis in B). High ploidy cells require more nutrients for growth (x-axis), setting high ploidy cells at a disadvantage when competing in nutrient-scarce micro-environments.
Our current research continues to explore how selective pressures like nutrient deprivation drive karyotype evolution through long-term experiments. We are investigating the hypothesis that nutrient stress causes CIN, creating a feedback loop where instability allows adaptation, reducing stress and thus CIN.

Long-term nutrient-deprivation drives convergent karyotype adaptation. Mueller plots track SNU-668 lineages under phosphate-deprived conditions. Three phosphate replicates converge on the same dominant clone (orange; gains of chr 11, 18 & 21), supporting the adaptive role of karyotype changes. Changes in growth rate and morphology (upper panels) are tracked in parallel.
Cell adaptations to new growth environments
...
Integrating live-cell imaging with single-cell sequencing
Exposing cancer cells to a new environment influences their growth behavior. For some cells, a moderate growth inhibition is followed by adaptation and return to normal growth rates. Others initially experience a near-complete cytostatic phenotype, only to explode in their growth during later generations, reaching growth rates well beyond baseline. This implies that one can reach opposite conclusions about the relative fitness of two cell lineages, solely depending on timing of measurement. Our goal is the development of a new class of temporal biomarkers that extrapolate from a cell's transcriptome how fit its descendants will be over multiple generations. We record how cells divide, migrate and die, linking the recorded phenotypic differences between cells to differences between their transcriptomes.
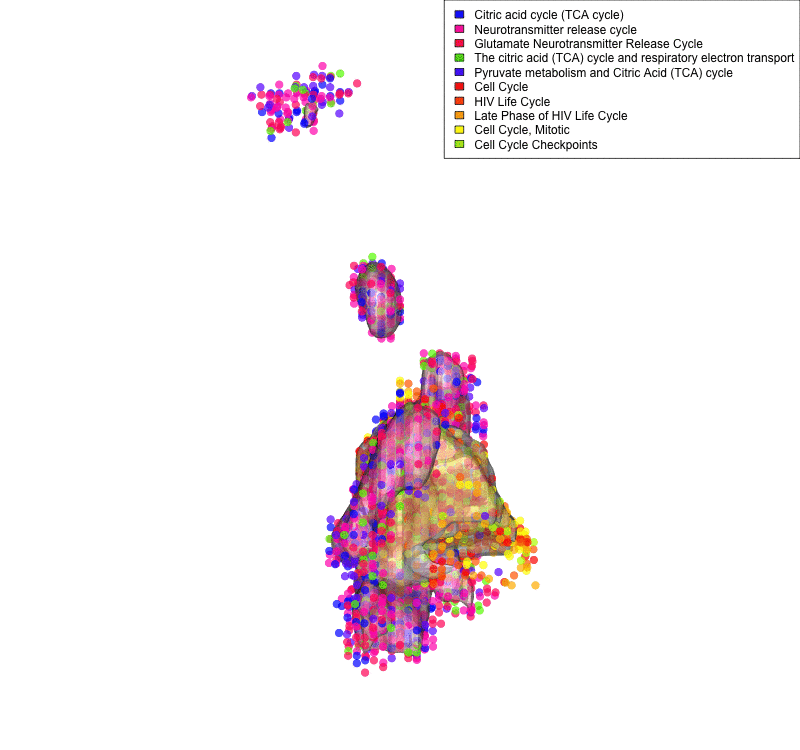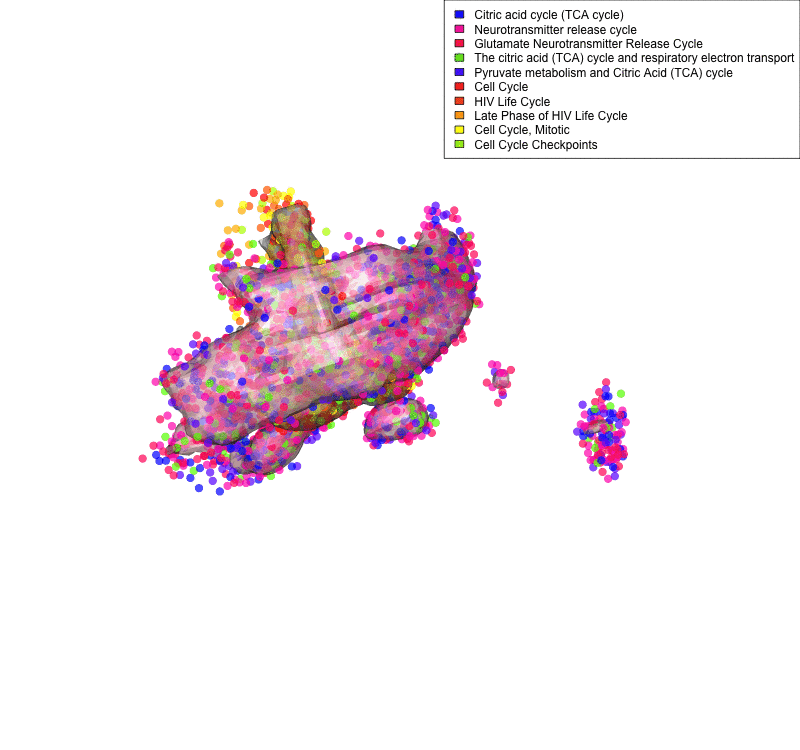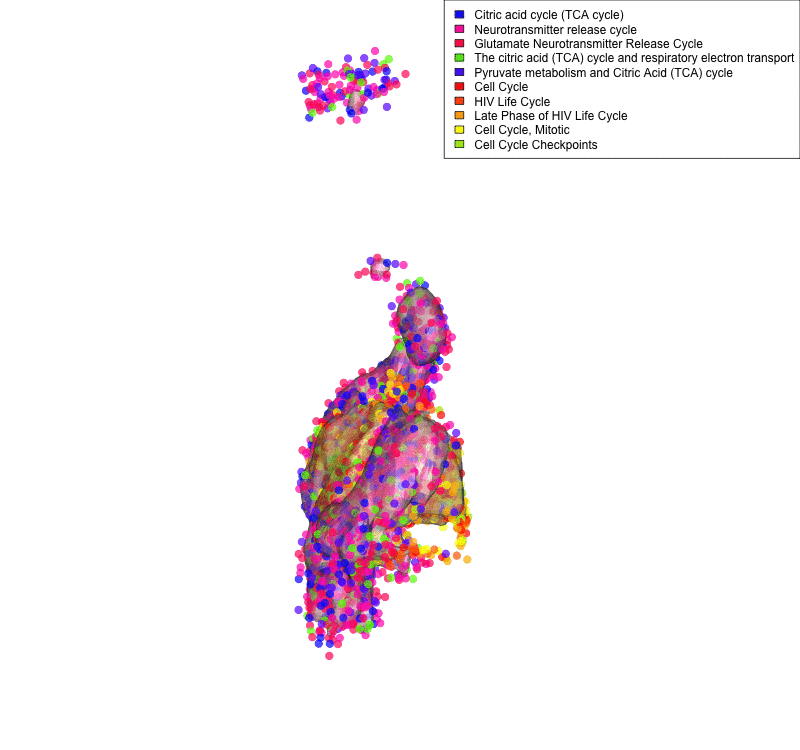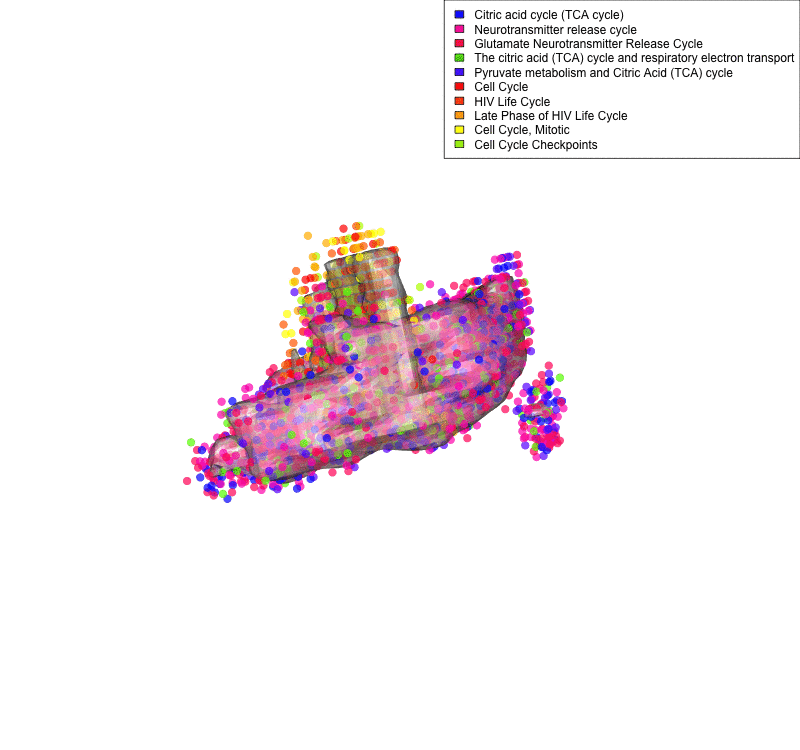
Linking sequenced and imaged cells allows visualizing the spatial segregation of pathways activity inside a cell (here shown for only two compartment: mitochondria and nucleus and 10 pathways: legend). Frequency of each color indicates strength of pathway activity.
We use live-cell imaging to characterize the cell cycle of cancer cell clones as they adapt to new environments. A four-layered approach maps sequenced- and imaged cells in-silico. Hereby biological variability – emerging from multiple growth conditions – acts as an additional barcode during sequencing. This linking matches the sequenced cell's transcriptome to its closest living relative still undergoing live-cell imaging. Integration with live-cell imaging opens the door to leverage the suitability of single cell sequencing for deep learning in a new way – not for solving technical challenges like segmentation and tracking, but for interpretation of genomic information.
Mathematical modeling of competition dynamics of co-existing tumor clones
The transcriptome is part of a cell's "vocabulary" that is used to communicate with its environment. As such, not only the fitness of a cell, but also its transcriptome, is context-dependent -- a function of environmental changes. Here we evaluate how environmental fluctuations – changes in nutrient availability and changes in the cellular population structure – affect the fitness of these subpopulations. If coexisting subpopulations interact with one another, then the selective forces acting on these subpopulations will no longer be constant, but will change with changes in the cellular fraction of each population.

Evolutionary game theory (EGT) is a mathematical framework designed to model this scenario of dynamic selection. We integrate single-cell sequencing into an EGT framework to restrict the search space of possible interactions between subpopulations and to characterize the nature (i.e., the strength and direction) of these interactions.